A newly developed nanobody could significantly expand treatment options for cystic fibrosis (CF). Researchers at Charité – Universitätsmedizin Berlin and the Leibniz-Forschungsinstitut für Molekulare Pharmakologie (FMP) have, for the first time, engineered a cell-permeable antibody fragment that acts directly inside cells. It stabilises and functionally improves the CFTR channel, which is frequently defective in cystic fibrosis. The results have been published in the journal Nature Chemical Biology.
In most cases, cystic fibrosis is caused by the so-called F508del mutation in the CFTR gene. This mutation leads to misfolding of the CFTR channel, causing it to be prematurely degraded within the cell. As a result, water and salt transport is disrupted, leading to the accumulation of thick mucus in the lungs, which promotes chronic infections and inflammation.
Modern CFTR modulators—particularly the triple therapy consisting of elexacaftor, tezacaftor, and ivacaftor—can already significantly improve channel function. However, not all patients respond sufficiently to this treatment, and full restoration of CFTR function is usually not achieved.
The newly developed nanobody directly targets the underlying defect: it binds to the misfolded CFTR protein within the cell and supports its correct folding. Using so-called cell-penetrating peptides, the antibody fragment can enter airway epithelial cells.
In cell models derived from cystic fibrosis patients, the nanobody was shown to stabilise the defective channel and restore its function. When combined with established triple therapy, a strong synergistic effect was observed: CFTR activity increased to approximately 90% of normal levels in experimental settings.
“Through this novel mechanism of action, CFTR function can be corrected much more effectively in combination with existing modulators,” explains Marcus Mall, Director of the Department of Paediatrics with focus on Pneumology, Immunology and Intensive Care Medicine at Charité. “Our results suggest that, in the future, a near-complete normalisation of CFTR function may even be achievable.”
However, further research is required before clinical application. Key challenges include the development of suitable inhalation-based formulations, as well as studies on in vivo efficacy and immunological compatibility.
Beyond cystic fibrosis, this approach may have broader relevance: intracellular nanobody-based therapies could open new avenues for treating diseases caused by protein misfolding, for which few effective therapies currently exist.
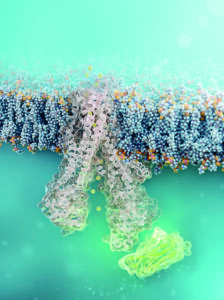
Cell-permeable nanobody (green) binds to defective CFTR chloride channel (structural simulation). © FMP | Barth van Rossum
Original publication: Franz L, Rubil T, Balázs A, Overtus M, Kemnitz-Hassanin K, Govaerts C, Mall MA, Hackenberger CPR. A cell-permeable nanobody to restore F508del cystic fibrosis transmembrane conductance regulator activity. Nat Chem Biol. 2026 Apr 17. doi: 10.1038/s41589-026-02199-w. Epub ahead of print. PMID: 41998105.



